



Next: redo_mlrgd and redo_plots
Up: usersguide
Previous: Error log file
Contents
Parameters
There are a variety of parameters that you can alter by opening the
script run_mlrgd in a text-editor and editing the Options
section. A brief description of these are given below (suggested
defaults are give by param = default).
- pairs = 1: Method for generating the list of sequence pairs to
use: 2
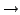 use phylogenetic tree built with ednapars (maximum
parsimony); 1 use phylogenetic tree built with ednaml
(maximum likelihood); 0 or other use user-input file
prefix.pairs.
use phylogenetic tree built with ednapars (maximum
parsimony); 1 use phylogenetic tree built with ednaml
(maximum likelihood); 0 or other use user-input file
prefix.pairs.
- refpos = 0: 0 use seperate CDS files for each
sequence; 1 or other use reference sequence CDS files for all
sequences.
- gbkpos = 0: 0 get CDS files from
seqname.fasta.orfs (or seqname.gbk.orfs) files; 1
or other get CDS files from seqname.gbk files
(for any seqname.fasta files, the seqname.fasta.orfs
file, if any, will be used).
- skip = 0: 1 or other skip sequence files
containing non-ACGTUacgtu characters (exits if the reference sequence
contains non-ACGTUacgtu characters); 0 read standard
ambiguous nt codes as N and ignore for most statistics (ambiguous nt
are logged along with the gaps; codons containing ambiguous nt are logged
along with the `zero-probability' transitions).
- window1 = 5:
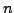 , where the running mean sliding window size
for the 1st, 2nd and 3rd codon positions and for the 4-fold degenerate
neutral sites plots is
, where the running mean sliding window size
for the 1st, 2nd and 3rd codon positions and for the 4-fold degenerate
neutral sites plots is 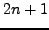 nt (or codons).
nt (or codons).
- window2 = 10: , where the running mean sliding window size
for the all sites and for the non-coding sites plots is nt.
- circular = 1: 0 not a circular genome; 1 or
other circular genome.
- clip1 = 0.00: Upper clipping threshold for clipped running
mean: clip1 = 0.05 means that the upper 5% (i.e. columns with low
observed number of mutations relative to expected number of mutations) in
each window are clipped before calculating the mean.
- clip2 = 0.30: Lower clipping threshold for clipped running
mean: clip2 = 0.05 means that the lower 5% (i.e. the columns with
high observed number of mutations relative to expected number of mutations)
in each window are clipped before calculating the mean.
- tttmin = 0.0: Lowest
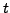 value to use in -fitting.
value to use in -fitting.
- tttmax = 10.0: Highest value to use in -fitting.
- tVfit = 0.2: Required fitting accuracy (total number of mutations
between a sequence pair) for and
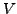 .
.
- maxiter = 20: Maximum number of fitting iterations for and .
- Vmin = 0.01: Lowest value to use in -fitting (
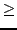 0.01).
0.01).
- Vmax = 10.0: Highest value to use in -fitting (
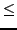 10).
10).
- Vtype = 1: 0 find seperate value for each
sequence pair; 1 or other find one value for the alignment.
- Vfix = 0: If
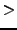 0, use this value for all sequence pairs
(defaults to 1 if 0; overridden if fitwhat = 3).
0, use this value for all sequence pairs
(defaults to 1 if 0; overridden if fitwhat = 3).
- fitwhat = 3: 0 or other fit using total number
of mutations; 1 fit using number of neutral/synonymous
mutations; 2 fit using number of neutral/synonymous
mutations at 4-fold degenerate sites; 3 as for 2, then adjust
to fit the total number of mutations (i.e. including nonsynonymous
mutations).
Note: 1--3 are not recommended if the sequence is predominantly double-coding
since, in this case, the number of neutral sites is potentially very small.
- wholeseq = 1: Nucleotide range to use for all model-fitting,
statistics and plots: 0 use the region
range1--range2, 1 or other use the whole sequence.
- range1 = 0: Start nucleotide of region to analyse
(if wholeseq = 0; otherwise ignored), in reference sequence coordinates.
- range2 = 0: End nucleotide of region to analyse
(if wholeseq = 0; otherwise ignored), in reference sequence coordinates.
Next: redo_mlrgd and redo_plots
Up: usersguide
Previous: Error log file
Contents
aef
2007-12-10