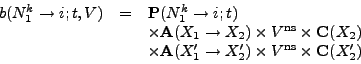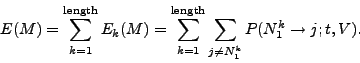In this section we describe the model for non-coding, coding, and multiply-coding regions. We broadly follow the standard approach of modelling sequence evolution as a Markov process (Goldman & Yang 1994; Hein & Stovlbaek 1995; Ewens & Grant 2001). However we do deviate a little (see below) in order to deal with potentially double-coding or triple-coding regions, without having to resort to time-consuming MCMC simulations to estimate probabilities (Pedersen & Jensen 2001). We use more or less the same approach as in Firth & Brown (2005, supplementary material).
Suppose we have an aligned pair of sequences ![]() and
and ![]() . For
each nucleotide
. For
each nucleotide ![]() in
in ![]() we estimate the probability that
we estimate the probability that
![]() mutates to each of the nucleotides U, C, A, G, as follows.
First we define
mutates to each of the nucleotides U, C, A, G, as follows.
First we define
![]() ,
,
![]() , by
, by
The probability that ![]() mutates to
mutates to ![]() , after time
, after time ![]() , is then
given by
, is then
given by
Using this model, we can calculated the expected number of non-null
mutations ![]() occurring in
occurring in ![]() after a given time
after a given time ![]() by
by
The parameter ![]() may be left at the default value of 1 (usually
pretty close to the best-fit value) or fitted seperately for each pair
of sequences or one value may be fitted for the whole alignment. In
the latter two cases, first
may be left at the default value of 1 (usually
pretty close to the best-fit value) or fitted seperately for each pair
of sequences or one value may be fitted for the whole alignment. In
the latter two cases, first ![]() is determined by fitting the observed
and expected number of mutations only at non-coding positions and
four-fold degenerate sites where the codons in
is determined by fitting the observed
and expected number of mutations only at non-coding positions and
four-fold degenerate sites where the codons in ![]() and
and ![]() are
synonymous. Then
are
synonymous. Then ![]() is determined by fitting the observed and
expected total number of mutations.
is determined by fitting the observed and
expected total number of mutations.
Any transition that is assigned a zero-probability according to the input amino acid and codon matrices (e.g. transitions between stop codons and non-stop codons in the default matrices) is ignored and written to the log file. Any transition involving gaps or ambiguous nucleotide codes is also ignored and written to the log file.
Note that the above methodology involves several approximations.
Firstly, in Equation 3, we consider only a single
nucleotide and the single codon in each of the primary and secondary
read-frames containing that nucleotide. However in double-coding
regions, adjacent codons are linked by the overlapping read-frame.
Hence mutations within one codon can affect the probabilities for
subsequent mutations in neighbouring codons. Secondly, as far as
codon and amino acid weightings are concerned, we consider only the
start and end points of the unknown mutation pathway connecting an
aligned codon pair in ![]() and
and ![]() . It seems reasonable that these
simplifications are justified provided
. It seems reasonable that these
simplifications are justified provided ![]() and
and ![]() are not too
divergent (so that mutation pathways are short and inter-codon
cross-talk is low in multiply-coding regions). In Firth & Brown
(2005) it is shown that this model provides useful results over a wide
range of circumstances.
are not too
divergent (so that mutation pathways are short and inter-codon
cross-talk is low in multiply-coding regions). In Firth & Brown
(2005) it is shown that this model provides useful results over a wide
range of circumstances.
The parameter ![]() models the selection pressure that may vary from one
gene to another. In fact the selection pressure will be different for
every codon and this is often modelled by site-dependent rates models
(Yang et al. 2000). Using a site-dependent model is pointless for
our purposes, as it will be impossible to distinguish columns that
have enhanced conservation. The BLOSUM amino acid matrix that we use
represents the average amino acid substitution acceptabilities
over all codons. The parameter
models the selection pressure that may vary from one
gene to another. In fact the selection pressure will be different for
every codon and this is often modelled by site-dependent rates models
(Yang et al. 2000). Using a site-dependent model is pointless for
our purposes, as it will be impossible to distinguish columns that
have enhanced conservation. The BLOSUM amino acid matrix that we use
represents the average amino acid substitution acceptabilities
over all codons. The parameter ![]() allows a global adjustment for
selection pressure while still allowing particularly conserved sites
to be distinguished from other sites. Such sites may be additional
non-coding elements, new coding ORFs, or more-conserved regions within
proteins (e.g. motifs). The track for mutations at four-fold
degenerate neutral sites may be used to help distinguish between these
alternatives, though it will provide information for less than one in
three nucleotides within CDSs. The track for 3rd codon positions may
also be used and, since it includes 2-fold degenerate sites, will
provide information at more positions.
allows a global adjustment for
selection pressure while still allowing particularly conserved sites
to be distinguished from other sites. Such sites may be additional
non-coding elements, new coding ORFs, or more-conserved regions within
proteins (e.g. motifs). The track for mutations at four-fold
degenerate neutral sites may be used to help distinguish between these
alternatives, though it will provide information for less than one in
three nucleotides within CDSs. The track for 3rd codon positions may
also be used and, since it includes 2-fold degenerate sites, will
provide information at more positions.